
We use the “Deep Potential” molecular dynamics (DPMD) approach of Car, E, and coworkers [Phys. Rev. Lett. 120:143001 (2018)] to develop force fields for fluids. The approach involves selecting a number (hundreds to thousands) of configurations for a system at thermodynamic state points representative of the conditions of interest. Kohn-Sham density functional theory is then used to obtain the “reference” energies of the corresponding configurations. For every configuration in the training set, a local coordinate frame is set up for every atom and its neighbors inside a smooth cutoff radius to allow for translational and rotational symmetries to be preserved. Permutational invariance is maintained by summing over all possible permutation of atoms of the same type. Then, these “descriptor” representations serve as inputs to a deep neural network to undergo linear and nonlinear transformations and output the energy for every atom. A molecular dynamics simulation is then run using the resulting potential energy surface, generating new configurations; their reference energies are evaluated. If differences between reference energies and the neural network model exceed specified thresholds, the network parameters are adjusted appropriately and the procedure is repeated.
A fundamental shortcoming of many machine learning models is that most such models only take into account interactions within a localized atom-centered spherical representation with a pre-defined radial cutoff. In a recent development, we have been developing methods for incorporation of long-range interactions into such models, as shown in the figure below.
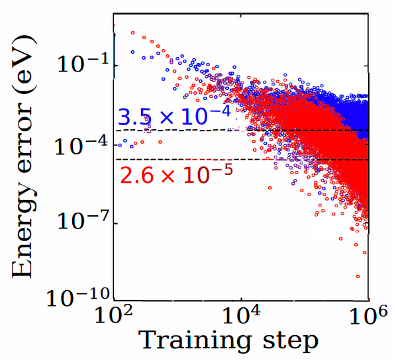
Training error in system energies for the finite-range (blue) and long-range (red) version of a model, from [J. Chem. Phys., 154, 034111 (2021). http://dx.doi.org/10.1063/5.0031215]