
Research activities in the Panagiotopoulos group focus on development and application of theoretical and computer simulation techniques for the study of properties of fluids and materials.
Emphasis is on molecular-based models that explicitly represent the main interactions in a system. These models can be used to predict the behavior of materials at conditions inaccessible to experiment and to gain a fundamental understanding of the microscopic basis for observed macroscopic properties.
Our work usually requires large-scale numerical calculations involving a number of powerful molecular simulation methodologies. An example of such a methodology is Gibbs ensemble Monte Carlo, which provides a direct way to obtain coexistence properties of fluids from a single simulation.
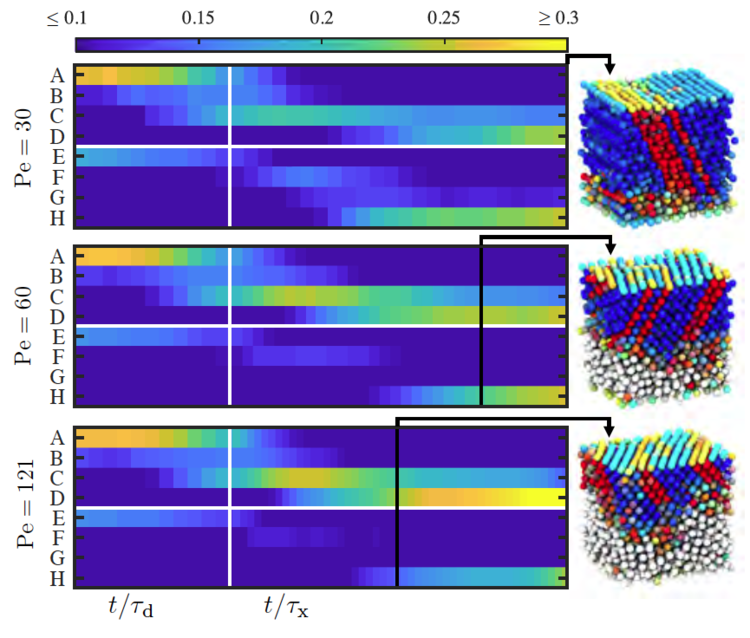
Time evolution of crystal structures for a solvent/particle system in which the solvent is evaporating at different rates [Howard et al., J. Chem. Phys. 149: 094901, 2018].